Трисомия 13, патау синдром
Содержание:
Синдром Патау – что это за болезнь?
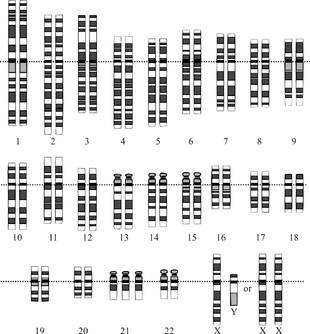
Синдром Патау характеризуется присутствием в клетках добавочной хромосомы номер тринадцать, т.е. вместо пары гомологичных хромосом данного типа присутствует три таких структуры. Аномалия также определяется термином «трисомия 13». В норме набор хромосом в клетках человеческого организма (нормальный кариотип) представлен 46 элементами (23 пары), из которых две пары отвечают за половые признаки. При исследовании кариотипа в клетках крови у любого человека могут быть выявлены изменения строения хромосом, не влияющих на его здоровье, но способных дать о себе знать у потомков.
Синдром Патау – тип наследования
При диагнозе «синдром Патау» кариотип записывается формулой такого вида: 47 XX (XY) 13+. При этом три копии тринадцатой хромосомы могут присутствовать во всех клетках тела, в других случаях дополнительная синтезированная хромосома встречается только в некоторых клетках. Это случается вследствие ошибки при делении клеток в начале развития зародыша после соединения яйцеклетки и сперматозоида, возникающей под действием каких-либо внутренних или внешних воздействий. Причем лишняя хромосома может исходить как от матери, так и от отца, не имеющих генетических отклонений.
Кроме этого, существуют случаи, когда дополнительная хромосома номер 13 может быть прикреплена к другой хромосоме в яйцеклетке или в сперматозоиде, что именуется транслокацией. Это является единственной формой синдрома Патау, которая способна передаваться от кого-то из родителей. Люди, являющиеся носителями измененного генетического материала и не имеющие признаков заболевания, могут передать его детям, которые родятся больными.
Риск трисомии 13

Синдром Патау у плода зачастую является горькой случайностью, от которой никто не застрахован. В последнее время многим парам рекомендуют провести кариотипирование перед планированием зачатия, даже если не установлен высокий риск синдрома Патау или прочих хромосомных аномалий. Эта методика изучает набор хромосом женщины и мужчины, выявляет различные отклонения. Как минимум, благодаря изучению генома родителей возможно спрогнозировать, есть ли вероятность наследственной формы патологии.
Как и многие другие хромосомные нарушения, рассматриваемая болезнь в большинстве случаев встречается у деток, зачатых женщинами старше 35-45 лет. Поэтому на ранних сроках беременности назначается, если есть высокий риск синдрома Патау, амниоцентез – исследование клеток плода на наличие генетических дефектов. Такой анализ осуществляется посредством пункционного прокола полости матки и забора околоплодных вод с наличием слущенных клеток плода.
Синдром Патау – частота встречаемости
Кариотип, свойственный синдрому Патау, регистрируется примерно один раз на каждые 7-14 тысяч новорожденных, появившихся на свет живыми. Встречаемость у мальчиков и у девочек одинаковая. Кроме того, беременности с таким отклонением у плода относятся к группе высокого риска выкидыша или мертворождения. В 75 % случаев родители малышей с данным диагнозом не имеют хромосомных отклонений, остальные эпизоды сопряжены с наследственным фактором – из-за передачи транслокализованой хромосомы номер 13 от одного из родителей.
Изменения внутренних органов
К сожалению, присутствие дополнительной хромосомы проявляется не только во внешности, страдает все тело. Поэтому при обнаруженных внешних признаках ребенку назначают полное обследование внутренних органов.
Центральная нервная система
У таких детей мозг имеет меньшие размеры, могут отсутствовать целые отделы. Либо они недоразвиты. Как правило, при этом заболевании страдает мозолистое тело, мозжечок. При такой аномалии ребенок поражен тяжелой формой идиотии. Он не способен говорить, мыслить, понимать, что вокруг происходит. Таким пациентам даже трудно контролировать свои движения. Поэтому они могут так и не научиться ходить, есть, и делать другие обыденные вещи. Данная патология не позволяет человеку чувствовать и выражать свои эмоции. Отсутствует восприимчивость к внешним раздражителям, например, яркому свету или громкому звуку.
Сердечно-сосудистая система
При синдроме Патау могут возникать аномалии сердечной перегородки, неправильное строение крупных сосудов, порок сердца, открытый артериальный проток.
Мочеполовая система
Почки при данном синдроме могут быть поражены поликистозом, подковообразной почкой, гидронефрозом. Мочеточник укорочен. У мальчиков – недоразвитость внешних половых органов, не опускается яичко в мошонку. У девочек – две матки, двойное влагалище, гипертрофия внешних половых органов.
Органы чувств
Недоразвитие мозга приводит к аномалиям формирования и функциональности органов зрения и слуха. Может присутствовать полная глухота. Отмечено рождение детей с данным синдромом, у которых один глаз был неполноценным или отсутствовал совсем. Кроме этого, проявляются и другие болезненные признаки: катаракта, дисплазия сетчатки и другие.
Другие проявления
Патология может вызывать перемену мест расположения органов, их дефектами и недоразвитостью. Кроме того при рождении наблюдается пупочная грыжа. На коже тоже могут быть патологические изменения, например, отсутствие волосяного или кожного покрова на участке тела, складки. Одним из опасных признаков болезни является лейкемия – рак крови.
Профилактика
Провести профилактику данного синдрома не представляется возможным – нельзя предугадать, что ребенок родиться с трисомией 18. Но, несмотря на наследственный характер заболевания, родители все же могут сделать кое-что для снижения риска возникновения заболевания у будущего ребенка.
В первую очередь, партнеры должны пройти полное обследование для выявления патологий в собственном организме.
Также следует обратить внимание на возраст, в котором предпринимаются попытки зачать ребенка – желательно, чтобы он не достигал 40 лет; в таком случае снизится риск возникновения прочих заболеваний у ребенка и осложнений у матери. И, пожалуй, один из главных аспектов, который еще не был затронут ранее, – правильное питание и исключение пагубного влияния вредных привычек на организм будущей матери
Лечение заболевания
Этиотропного лечения для хромосомных болезней не существует, в случае с синдромом Шерешевского Тернера симптомы можно только облегчить с помощью гормон заместительной терапии. Применяется гормон роста, в период начала полового созревания используют препараты эстрогена и эстрадиола, а после назначают оральные прогестин содержащие контрацептивы с целью восполнить уровень не достающих организму гормонов. Такие часто встречающиеся при хромосомной патологии проблемы как аномалии развития сердца и сосудов при наличии показаний лечат оперативно. Для снижения явлений застоя лимфы при лимфедеме назначают массаж, специальные физические упражнения а также компрессионные чулки.
Заболевание отличается благоприятным для жизни прогнозом (при условии отсутствия, или своевременной коррекции врождённых пороков сердца и магистральных сосудов), для деторождения же прогноз неблагоприятный – большинство пациенток не в состоянии забеременеть.
Причины синдрома Патау
Установлено, что причиной синдрома Патау, в большинстве случаев, является утроение 13 хромосомы, то есть в каждой клетке имеется не две (норма), а три копии тринадцатой хромосомы. В очень редких случаях только часть клеток организма имеет дополнительную копию. Это так называемый мозаичный синдром Патау.
Еще одна причина заболевания – транслокация (перестройка) хромосом, когда часть 13 хромосомы до или непосредственно в момент зачатия привязывается к другой негомологичной хромосоме. В результате такой перестройки у больных наряду с двумя копиями 13 хромосомы появляется дополнительный материал из нее, который подключен к другой хромосоме. Возникает частичная трисомия 13 хромосомы, при которой физические признаки синдрома несколько отличаются от типичной клинической картины.
Как правило, синдром Патау не наследуется, а возникает случайно в процессе формирования сперматозоидов и яйцеклеток. Если при делении клеток возникает ошибка, которая называется нерасхождением, это приводит к появлению половых клеток с неправильным числом хромосом. Когда подобные атипичные сперматозоиды и яйцеклетки вовлекаются в генетическую структуру ребенка, он получает дополнительную 13 хромосому во всех клетках организма. Мозаицизм данного синдрома также не наследуется и возникает как случайный сбой при делении клеток на первоначальном этапе развития зародыша.
Заболевание может быть унаследовано в случае сбалансированной транслокации, когда здоровый человек несет измененный генетический материал между тринадцатой и другими хромосомами. Лица, являющиеся носителями, входят в группу риска по рождению детей с этим генетическим заболеванием, хотя сами и не имеют признаков трисомии хромосомы 13.
Синдром Патау
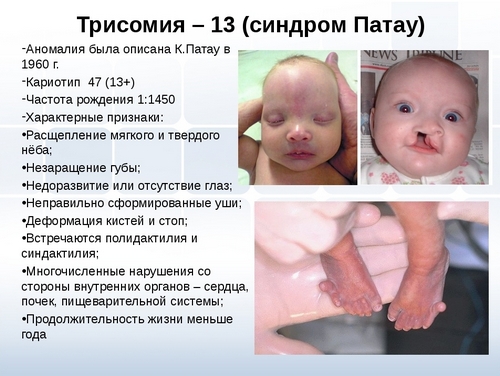
Синдром Патау – хромосомное заболевание, обусловленное наличием дополнительной копии 13-ой хромосомы (трисомия по 13-ой хромосоме).
В структуру синдрома Патау входят множественные дефекты нервной системы (микроцефалия, голопрозэнцефалия), глаз (микрофтальмия, катаракта), костно-мышечной системы (полидактилия, расщелины губы и нёба, омфалоцеле), сердца, урогенитальной системы и др.
Для выявления и подтверждения синдрома Патау проводится пренатальный скрининг, исследование кариотипа ребенка после рождения. Детям с синдромом Патау необходима общеукрепляющая терапия; по показаниям – коррекция врожденных пороков развития.
Синдром Патау — хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13. Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое.
Клинический симптомокомплекс был описан еще в XVII веке; связь же заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название.
При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией.
Диагностика синдрома Патау
Пренатальная диагностика хромосомных болезней плода (синдрома Патау, синдрома Дауна, синдрома Эдвардса) одинакова. На первом этапе скрининга производится определение биохимических маркеров (бета-ХГЧ, РАРР-А и др.) и УЗИ-исследование, на основании которых рассчитывается риск рождения больного ребенка для данной женщины.
Женщинам, попавшим в группы риска, предлагается проведение инвазивной пренатальной диагностики: биопсии ворсин хориона (8-12 недели), амниоцентеза (14-18 недели) или кордоцентеза (после 20-й недели гестации). В полученных образцах материала плода проводится поиск трисомии по 13-ой хромосоме методом кариотипирования с дифференциальной окраской хромосом или КФ-ПЦР.
Новорожденные с предполагаемым или установленным диагнозом синдрома Патау нуждаются в углубленном комплексном обследовании для выявления тяжелых пороков развития (эхокардиографии, УЗИ органов брюшной полости и почек, нейросонографии, КТ головного мозга и др.). Для определения показаний к оперативному лечению, в первую очередь, необходимы консультации детского кардиохирурга и детского хирурга общего профиля.
Возможности медицинской помощи детям с синдромом Патау ограничены и сводятся, главным образом, к организации хорошего ухода, полноценного питания, профилактике инфекций, общеукрепляющей и симптоматической терапии. Хирургическая помощь может потребоваться для устранения врожденных пороков сердца, расщелин лица и др.
Дети с синдромом Патау находятся под наблюдением педиатра, детского генетика, детского невролога, детского кардиолога, детского офтальмолога, детского травматолога-ортопеда, детского отоларинголога, детского гастроэнтеролога, детского уролога и других специалистов.
Прогноз и профилактика синдрома Патау
В большинстве случаев плод синдромом Патау погибает антенатально или рождается мертвыми. Живорожденные дети также имеют неблагоприятный прогноз для жизни. В большинстве случаев продолжительность их жизни не превышает одного года.
Специфические методы профилактики синдрома Патау не разработаны. При наличии хромосомных заболеваний в предыдущих поколениях или случаев мертворождения перед планированием беременности родителям необходимо пройти медико-генетического консультирование.
Прогноз
Если женщина всё же выберет продолжение беременности, или в том случае, если аборт будет уже противопоказан по срокам, то за исключением редких случаев придётся отказаться от прерывания
К тому же, должно быть сосредоточено серьёзное внимание на том, как именно стоит рожать
Плод с синдромом Патау в большинстве случаев погибает антенатально (во время беременности) или рождается уже мёртвым. Это объясняется значительной тяжестью врождённых аномалий внутренних органов. Малыши с данной патологией, которые после рождения остаются в живых, всю свою жизнь являются полными инвалидами и страдают идиотией.
Если причиной заболевания стал случайный фактор, то последующие беременности у этой женщины могут быть успешными.
В большинстве случаев плод синдромом Патау погибает антенатально или рождается мертвыми. Живорожденные дети также имеют неблагоприятный прогноз для жизни. В большинстве случаев продолжительность их жизни не превышает одного года.
Тем не менее, в странах с высоким уровнем медицины есть дети с трисомией 13 хромосомы, которые прожили до 10 лет. Однако, сильная умственная отсталость, операции и сложнейшее лечение не дадут эффективного результата, а лишь продлят срок жизни.
Ребенок с этим заболеванием никогда не будет полностью самостоятельным и независимым. Из-за сильной умственной отсталости детям с синдромом Патау сложно адаптироваться в обществе, строить взаимоотношения.
Согласно статистике, приблизительно один ребёнок из 150 рождается с какой-либо хромосомной аномалией. Такие нарушения возникают в результате ошибок в структуре или количестве хромосом. Многие дети с подобными проблемами имеют физические и/или психические врождённые аномалии. Некоторые из них в конечном итоге провоцируют выкидыш или даже мертворождение. Одним из наиболее часто стречаемых является синдром Патау.
Предлагаем ознакомиться Признаки рака яичников у женщин и первые симптомы заболевания
Хромосомами называются нитевидные структуры, которые находятся во всех клетках человеческого организма и содержат в себе определённый набор генов. В среднем у людей насчитывается порядка 20 — 25 тысяч генов, определяющих, например, цвет глаз или волос, а также отвечающих за развитие и рост всех частей тела.
Синдром Патау — это генетическое заболевание, представляющее собой утроение хромосом по XIII паре (так называемая Трисомия 13). Он приводит к многочисленным врождённым заболеваниям нервной системы, мышечной ткани и других систем и органов человека, которые зачастую провоцируют гибель ребёнка ещё во внутриутробном периоде и значительно ограничивают продолжительность его жизни, если он всё-таки рождается.
К сожалению, каких-то мер профилактики, направленных на предотвращение синдрома Патау, на сегодняшний день не разработано. Однако можно применять некоторые полезные рекомендации, с помощью которых удастся уменьшить риск появления недуга:
- переехать в другую местность, если она отличается плохой экологией;
- не взаимодействовать с химичесскими вредными компонентами;
- не устанавливать близкие интимные связи с кровными родственниками;
- планировать пополнение в семействе до 45 лет.
На фоне тяжелых врожденных аномалий развития исход данного заболевания крайне не благоприятен. Оно способно привести к таким последствиям:
- самопроизвольные аборты;
- большие риски появления на свет мертвого ребенка;
- около 95 % детей погибают сразу после рождения.
Если так случилось, что вы стали родителем особенного ребенка, не стоит огорчаться и падать духом. Вам следует искать единомышленников, общаться с родителями, которые также воспитывают особенных деток.
Учитывая все сказанное в этой статье, можно сделать вывод, что данная мутация всерьез способна усложнить жизнь малыша. Если взрослые не решились на аборт либо по какой-то из причин не удалось установить правильный диагноз при беременности, тогда нужно приложить колоссальное количество сил, чтобы продлить жизнь ребенка.
В большинстве случаев плод синдромом Патау погибает антенатально или рождается мертвыми. Живорожденные дети также имеют неблагоприятный прогноз для жизни. В большинстве случаев продолжительность их жизни не превышает одного года.
Симптомы
Первые клинические признаки патологии обнаруживаются уже при беременности. Срок ее сокращается до 38 недель, а течение осложняется многоводием. Врожденные аномалии, формирующиеся при синдроме Патау, обычно приводят к гибели плода внутри утробы матери. Выкидыши и мертворождение — самый частый исход патологии.
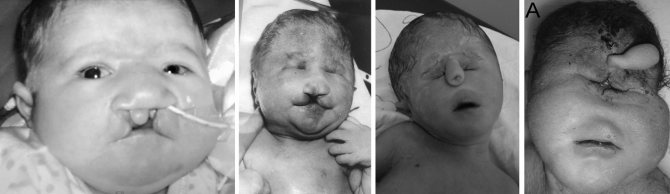
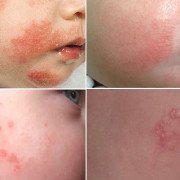
синдром Патау различной тяжети
Нередко в процессе родов младенцы задыхаются. Они рождаются с очень малой массой тела, так называемой пренатальной гипотрофией. Отличительные признаки заболевания у больных детей — симптомы поражения костно-суставного и мышечного аппарата, ЦНС, органа зрения, внутренних органов.
- Особенности внешнего вида: килевидная деформация черепа, микроцефалия, атипичный размер и форма лба, близко посаженные орбиты, сужение глазной щели, широкий нос, низкие ушные раковины, вогнутая переносица, укороченный шейный отдел позвоночника. Больные рождаются с серьезными дефектами лица — расщелиной неба и верхней губы. Подобные уродства делают больного ребенка не похожим на человека.
- Признаки поражения костей, суставов, мышц: сжатая в кулак кисть, полидактилия, сращение пальцев, деформированная стопа.
- Симптомы со стороны ЦНС: недоразвитие мозжечка, водянка головного мозга, дисгенезия мозолистого тела, грыжи различных отделов позвоночника, глухота.
- Врожденные аномалии зрительного анализатора проявляются колобомой, помутнением роговицы, микрофтальмией, врожденным помутнением хрусталика, атипичным строением сетчатки, недоразвитием зрительного нерва и его гипофункцией, отсутствием одного глаза, циклопией.
- Эндокринные нарушения — гипопитуитаризм.
- Поражение внутренних органов: пищеварения – воспаление и гетеротопия поджелудочной железы, кисты в железистой ткани, незавершенный поворот кишечника, дивертикул Меккеля, наличие добавочной селезенки; мочевыделения – увеличение почки и ее водянка, капиллярные гемангиомы и поликистоз почек, недоразвитие или дублирование мочеточника, отсутствие задней стенки уретры; сердца и сосудов — нарушение строения сердца, дефекты его перегородок, изменение нормальных объемов крупных и средних сосудов, разобщение малого и большого круга кровообращения; половых органов – крипторхизм, гипертрофия клитора, двурогая матка, недоразвитый половой член. Аномально развитая половая система — причина полной утраты репродуктивной способности, не подлежащей восстановлению.
- Младенцы нередко страдают от грыжи пупка, имеют глубокую степень умственной отсталости и входят в группу риска по развитию лейкоза.
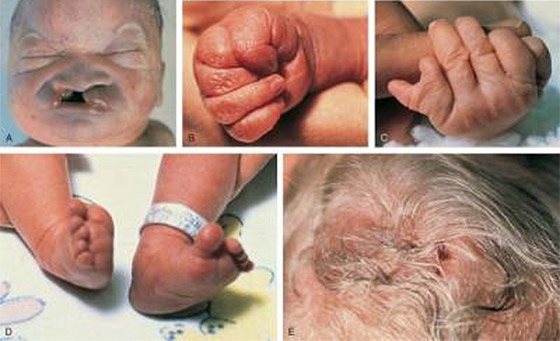
Новорожденные с такими патологиями редко доживают до года: 95% больных умирает практически сразу после родов. Продолжительность жизни при данном синдроме в отдельных случаях может достигать пяти или десяти лет.
На основании клинических проявлений болезни и особенностей течения патологического процесса выделяют следующие формы синдрома:
- Легкая форма — ребенок рождается с особенностями внешнего вида, отличающими его от остальных детей, гипотрофией и незначительными функциональными нарушениями внутренних органов. Продолжительность жизни колеблется от нескольких месяцев до года. При отсутствии тщательного ухода и медицинского наблюдения болезнь быстро прогрессирует.
- Умеренная форма – новорожденный имеет характерный внешний вид, обезображивающий его и позволяющий поставить предварительный диагноз сразу после рождения, а также дисфункцию жизненно важных органов, нарушающую процессы жизнедеятельности. Больные дети в среднем живут 2-3 дня и погибают от развившихся осложнений. Продлить жизнь таким детям поможет только хирургическое вмешательство, устраняющее врожденные аномалии.
- Тяжелая форма — ребенок погибает внутриутробно или умирает спустя несколько часов от рождения от полиорганной недостаточности.
Причины возникновения
Ребёнок с синдромом Патау является абсолютной случайностью для его родителей. Он может появиться в семье, в которой все члены могут похвастаться превосходным уровнем здоровья.
Трисомия
Как было уже сказано выше, основная причина возникновения данной аномалии — трисомия по XIII паре хромосом. Таким образом, с этим синдромом ребёнок имеет дополнительную 13-ю хромосому.
Ещё одна причина этого заболевания — это перестройка (так называемая транслокация), когда часть тринадцатой хромосомы перед зачатием или непосредственно в его момент привязывается к другой, в результате чего наряду с двумя копиями возникает дополнительный материал из неё, подключённый к другой хромосоме. Образуется частичная трисомия, при которой признаки синдрома немного отличаются от обычной клинической картины.
Синдром Патау, как правило, не наследуется, а появляется совершенно случайно в процессе образования яйцеклеток и сперматозоидов. Если во время деления клеток происходит ошибка в виде нерасхождения, то это приводит к формированию половых клеток, имеющих неправильное число хромосом. Когда такие атипичные яйцеклетки и сперматозоиды вовлекаются в структуру генов ребёнка, он получает ещё одну XIII хромосому во всех клетках своего организма.
Данное заболевание может быть также унаследовано вследствие сбалансированной перестройки, когда абсолютно здоровый человек имеет изменённый генетический материал. Лица, которые являются такими носителями, относятся к группе риска по рождению у них детей с синдромом Патау, хотя сами при этом не имеют признаков трисомии 13 хромосомы.
Факторы риска
Следует отметить факторы риска, которые способствуют развитию данного заболевания:
- дети, рождённые от браков между близкими родственниками;
- плохая экологическая обстановка в районе проживания;
- возраст матери более 45 лет;
- наследственные заболевания в предыдущих поколениях родителей.
Для данной болезни характерна абсолютная спонтанность, и предотвратить её нельзя.
Симптомы и признаки
Существуют разные признаки синдрома Патау, которые не спутаешь ни с какой другой болезнью. Так как это геномная мутация, её симптомы ярко выражаются даже во внешности. Врождённые пороки нередко приводят к летальному исходу детей ещё в младенчестве. Так как налицо внешние уродства, родители часто отказываются от таких деток. Какие симптомы синдрома Патау обычно диагностируются?
Патологии беременности
Явные УЗИ-признаки синдрома Патау:
- внешние уродства;
- замедленное развитие плода;
- тахикардия (в 70% случаев);
- многоводие (в 50% случаев) — этот признак позволяет выявить синдром Патау на УЗИ с учётом других диагностик;
- мегацистис (увеличенный в размерах мочевой пузырь);
- голопрозэнцефалия — неразделение мозга на два полушария;
- омфалоцеле — эмбриональная пуповинная грыжа.
Внешние симптомы
- Масса тела ниже нормы (менее 2 500 гр);
- умеренная микроцефалия — уменьшенные размеры черепа и головного мозга;
- низкий лоб, чаще всего скошенный;
- узкие глазные щели на небольшом расстоянии друг от друга;
- микроофтальмия — недоразвитие глаза;
- колобома — отсутствие глазной оболочки;
- запавшая переносица;
- помутнение роговицы;
- деформированные ушные раковины;
- расширенное основание носа;
- расщелины нёба или верхней;
- полидактилия — большее количество пальцев на руках и ногах, чем у других людей;
- изгиб кистей;
- короткая шея.
Внутренние симптомы
- Нарушение функционирования отделов ЦНС;
- пороки развития сердца (в 80%): дефекты перегородок (межжелудочковой и межпредсердной), транспозиции сосудов;
- добавочные селезёнки;
- фиброкистозные изменения в поджелудочной железе;
- эмбриональная пупочная грыжа;
- увеличенные почки;
- дольчатость почек;
- наличие в корковом слое почек кисты;
- патологии половых органов: их гипоплазия, у мальчиков крипторхизм — неопущение яичек, у девочек — двурогая матка;
- задержка умственного развития;
- отсутствие задней стенки мочеиспускательного канала.
Основные, типичные признаки заболевания выявляются при УЗИ уже на 12 неделе беременности, а затем подтверждаются всевозможными диагностическими исследованиями. Если они не проводились пренатально, клиническая картина синдрома Патау после рождения малыша достаточно ярко иллюстрирует именно данный диагноз. Но для исключения ошибки проводятся анализы на кариотип. В генетике различают несколько типов патологии.
Клиническая картина синдрома
Обычно дети с синдромом Шерешевского-Тернера, рождаются раньше срока. Но даже в случае беременности, доношенной до 40 недель, вес новорожденного редко достигает 3 кг, а рост более 50 см. Сразу же после родов, можно заметить у ребенка признаки, которые характерны для заболевания:
- Укороченная шея;
- Птеригиум-синдром – кожные складки в виде крыльев на боках шеи;
- Нарушение оттока лимфы;
- Отеки на стопах и кистях;
- Наличие врожденных пороков сердечно-сосудистой системы.
Далее на первый план выступают проблемы с кормлением – у детей нарушено сосание, они часто срыгивают «фонтаном», находится в моторном возбуждении. На первых годах жизни можно заметить отставание в развитие – ребенок поздно начинает сидеть, ходить, говорить. Также для синдрома Шерешевского-Тернера характерно частое, повторное возникновение среднего отита, которое впоследствии приводит к кондуктивной тугоухости.
На момент полового созревания люди с синдромом Шерешевского-Тернера выглядят следующим образом: рост людей редко когда превышает 150 см. Кроме этого, они имеют характерный внешний вид:
- Лицо обретает определенное выражение – «лицо Сфинкса»;
- Шея укорочена, присутствует птеригиум-синдром;
- Граница роста волос занижена;
- Челюсть недоразвита – микрогнатия;
- Уши увеличены, часто бывает лопоухость;
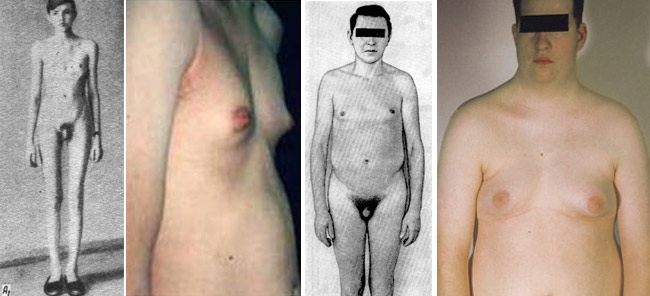
- Грудная клетка слишком широкая.
Жизнь с синдромом Шерешевского-Тернера непроста, при данной патологии значительно поражается костная система организма. Часто возникает сколиоз, дисплазия суставов, особенно тазобедренных, девиация локтей. На черепе наблюдается микрогнатия, неправильный прикус и готическое небо. В связи с недостаточным количеством эстрогена, люди с таким синдромом подвержены раннему возникновению остеопороза. У них часто случаются переломы позвоночника, костей кисти и шейки бедра.
Сколько живут люди с синдром Шерешевского-Тернера зависит от степени тяжести заболевания и от осложнений систем органов, которые возникают вследствие данной патологии . Со стороны сердечно-сосудистой системы встречаются такие пороки, как коарктация и аневризма аорты, дефекты межжелудочковой и межпредсердной перегородки. В почках часто бывает раздвоение лоханок, стеноз артерий, подковообразная форма самой почки. Подобные патологии приводят к артериальной гипертензии. У больных синдромом Шерешевского-Тернера нередко развивается косоглазие, близорукость и опущение века. К тому же они могут также страдать и дальтонизмом.
Умственные способности, как правило, сохраняются на должном уровне, но в некоторых случаях может наблюдаться олигофрения. Людей, страдающих синдромом Шерешевского-Тернера, часто сопровождают соматические заболевания – алопеция, микседема, витилиго, дефицит ферментов в тонком кишечнике, ожирение. Практически во всех случаях диагностируется диабет первого или второго типа и ишемическое поражение сердца. Доказано, что у таких пациентов рак толстого кишечника возникает в несколько раз чаще, чем у здоровых людей.
Практически у 100% больных девушек диагностируется первичный гипогонадизм. Внутренние половые органы недоразвиты – матка гипоплазирована, а вместо яичников с двух сторон находятся фиброзные тяжи. Женщины с синдромом Шерешевского-Тернера обычно полностью стерильны – в яичниках отсутствуют фолликулы с яйцеклетками. Вульва видоизменена: большие губы похожи на мошонку, а малые и клитор недоразвиты. Девственная плева может отсутствовать. Некоторые задаются вопросом как приходят месячные с синдромом Шерешевского-Тернера – в период полового созревания обнаруживается первичная аменорея. Грудные железы развиты недостаточно, наблюдается незначительное оволосение в участке лобка и подмышек. Беременность, которая возникла природным путем, возможно только в случае мозаичной формы поражения X-хромосомы.
Половые органы мужчины также недоразвиты. Яички гипоплазированы, часто не опускаются в мошонку, иногда диагностируется анорхия – полное отсутствие ткани яичек в организме. Наблюдается сильно заниженный уровень мужского гормона – тестостерона.
Диагностические мероприятия
Диагностика данной патологии, как и любого другого генетического заболевания, проводится пренатально и постнатально. Внутриутробное обнаружение подобных аномалий позволяет сопоставить возможные риски рождения больного ребенка. Методы пренатальной диагностики делятся на инвазивные и неинавазивные.
Неинвазивные методы считаются безопасными. Чтобы обнаружить генетическую аномалию, не требуется материал плода. Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока рекомендованы всем беременным женщинам. Диагностика с помощью УЗИ является достоверной на 100%.
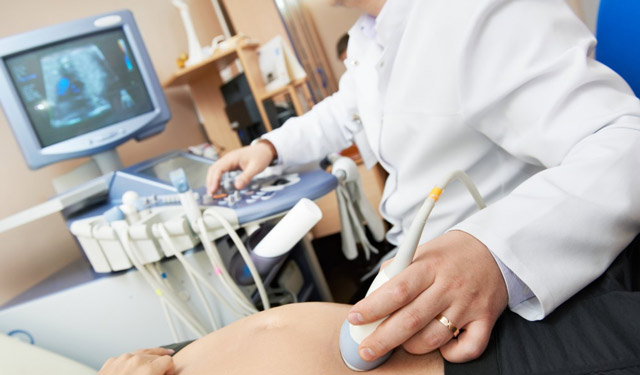
Стандартный пренатальный скрининг включает анализ крови на биохимические маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. Если полученные данные выходят за пределы нормальных значений, врачи рекомендуют прервать беременность. Для анализа необходима венозная кровь беременной женщины, в которой имеются фрагменты генетической структуры плода.
Инвазивные методы включают:
- Биопсию ворсин хориона, позволяющую диагностировать недуг с 8 по 12 неделю беременности. Одну из плодных оболочек берут на анализ с помощью пункционной иглы. Для исследования достаточно небольшого количества материала.
- Амниоцентез — забор околоплодных вод специальной иглой через брюшину для проведения клеточного анализа. Метод актуален с 14 по 18 неделю беременности. Исследование проводится под контролем УЗИ. Клетки, содержащие ДНК плода, проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пуповинной крови плода, позволяющее определить генетические аномалии с высокой точностью. Проводится оно после 20-й недели гестации.
Несмотря на высокую точность и надежность инвазивных методов, их применяют только в крайних случаях. Внедрение в организм беременной женщины и проникновение в оболочки плода несет определенный риск. Любое неверное движение специалиста, проводящего процедуру, может привести к внутриутробной гибели плода.
Диагноз устанавливают после определения кариотипа младенца путем КФ-ПЦР. Только анализ ДНК позволяет подтвердить или опровергнуть предполагаемый диагноз. С помощью хромосомных исследований можно определить, есть ли у ребенка трисомия 13, наследственная ли у аномалии этиология или она обусловлена спонтанной внутриутробной мутацией. Чтобы оценить риски и суметь предотвратить подобные мутации, родители должны пройти подробное генетическое исследование, которое дает 100%-ный результат. Генетическая экспертиза проводится, даже если произошла внутриутробная смерть плода, с целью предотвращения синдрома при повторной беременности.
Постнатальная диагностика заключается в обследовании новорожденного неонатологом и выявлении яркой и специфичной симптоматики. Больным детям показано тщательное всестороннее обследование, включающее следующие инструментальные методики:
- Кардиографию – электрокардиографию, фонокардиографию, коронарографию, магнитокардиографию;
- УЗИ внутренних органов, расположенных в абдоминальной полости и малом тазу: поджелудочной железы, почек, мочеточников, органов репродуктивной системы;
- Нейросонографию — УЗИ головного мозга;
- Томографическое исследование мозговых структур — КТ или МРТ.
Больным детям обычно требуется консультация врачей — специалистов в области офтальмологии, отоларингологии, неврологии, детской хирургии, генетики, эндокринологии, психиатрии.