«нога аиста» и ещё 7 признаков болезни шарко-мари-тута
Содержание:
- Болезнь Шарко причины
- Возможные осложнения
- Историческая справка
- Как проявляется болезнь Шарко-Мари-Тута?
- Факторы риска и причины ШMT
- Первый тип
- Что делать, если обнаружено заболевание
- Лечение болезни Шарко-Мари-Тута
- Историческая справка
- Лечение
- 1.2.2. Невральная амиотрофия шарко-мари-тута.
- Лечение БАС
- В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
Болезнь Шарко причины
Боковой амиотрофический склероз (болезнь Шарко) был открыт ещё в 1847 году, а описание клинических симптомов относится к 1869 году. Но наиболее активно это заболевание нервной системы стали изучать в 90-е годы XX столетия.
Болезнь Шарко считается семейной патологией, хотя известны случаи и спорадического характера, которые доказали, что основной причиной бокового амиотрофического склероза считаются мутации, происходящие в гене супероксиддисмутазы. В основном такие мутации были обнаружены в 20% семей с данным диагнозом, а вот роль этого гена в спорадических мутациях ещё до конца не определена.
Болезнь Шарко относится к самым частым прогрессирующим патологиям мотонейронов и является одним из наиболее тяжёлых дегенеративных аномалий центральной нервной системы. Установлено, что женщины в Америке и Европе болеют этим заболеванием гораздо реже, чем мужчины. А случаи семейной этиологии встречаются в 5–10% случаев, поэтому в основном характерна спорадическая картина болезни.
В развитии болезни Шарко, как и других заболеваний, при которых обнаруживаются поражения двигательных нейронов, главной причиной считаются также инфекционно-токсические заболевания, вызываемые определённым вирусом, который обладает большой нейротропностью. Кроме этого, болезнь Шарко развивается, как правило, в 50 лет.
Возможные осложнения
Лечение болезни Шарко хоть и не приводит к полному выздоровлению, но все же улучшает качество жизни пациента. Однако в ряде случаев, если поражаются нервы, отвечающие за диафрагму, дыхание может быть затруднено. В такой ситуации используются уже бронхолитики, вплоть до искусственной вентиляции легких.
У лиц с данной патологией есть большой риск получить травму. Пациентам лучше отказаться от кофе, алкоголя и табакокурения. В повседневной жизни лучше остерегаться стрессов и беречься от депрессий. Все эти факторы только ухудшают состояние больного. В этом случае может помочь когнитивная поведенческая терапия.
Последние стадии заболевания приводят к полной атрофии мышц, больной утрачивает полностью способность совершать движения, а это прямой путь к параличу и, как следствие, – летальный исход.

Историческая справка
Впервые официальное описание заболевания появилось в 1886 году в журнале La revue de medicine. Одним из авторов был Жан Мартен Шарко (Франция), которому на тот момент уже шел 61 год и он был известным медиком страны. Вторым автором, участвующим в описании заболевания и его симптомов, был его ученик, который практиковал всего лишь 3 года и звали его Пьер Мари.
Вторая статья была написана совсем молодым неврологом Говардом Генри Тутом (Англия), он только год назад получил приставку к своему имени – MD. И с того момента, заболевание получило название, состоящее из фамилий трех врачей.
Понятное дело, что это были не первые публикации о перонеальной мышечной атрофии. Еще ранее в записях Рудольфа Вирхова (1855 год) присутствует описание заболевания, но не столь подробное и без признаков.
Как проявляется болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.

Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы. Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе. Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте. Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног. Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При ШMT 1
мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга. Это приводит к мышечной слабости и потере чувствительности или онемению.
При ШМТ 2
мутирующий ген влияет непосредственно на аксоны. Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
ШМТ 3
илиболезнь Дежерин-Соттас , редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4
является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х
вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.
Первый тип
Данная разновидность присуща пациентам в возрасте 10-20 лет. Симптомы Шарко в данном случае изначально в слабости в области голеней. Начинается атрофия, и пациенту тяжело приподнять стопы, такое проявление называют еще «свисающая стопа». Чуть позднее появляется слабость в кистях рук, утрачивается чувствительность, появляются болевые ощущения.
Слабовыраженный тип характеризуется молоткообразными пальцами и высоким сводом стопы. Чаще всего этот тип ярче выражен у мужской половины человечества. Зато болезнь достаточно долго развивается, поэтому никак не влияет на продолжительность жизни.
Что делать, если обнаружено заболевание
Лечение помогает улучшить состояние и замедлить развитие невральной амиотрафии. Но всё равно необходимо будет придерживаться некоторых правил и вести определенный образ жизни. Нужно будет почаще делать физические нагрузки. Если сразу же начать их выполнять, то это быстрее принесет положительный эффект. Человек, который болеет данным заболеванием, ему необходимо выбрать такой вид спорта, чтобы были незначительные нагрузки.
Это может быть велосипед, плавание, катание на лыжах. Работа также должна быть специально подобрана под пациента и без сильных физических напряжений. Обувь обязательно должна быть удобной и не давить. Если у больного уже имеется «свисающая стопа», то ему нужно будет подбирать себе туфли под заказ, чтобы там был специальный фиксирующий ортез. Если обувь будет правильная, то это поможет избежать падения и травмы.
Обязательно питание должно быть правильным, это дает возможность избежать лишних килограмм. Ведь ожирение приносит дополнительную нагрузку на слабые мышцы.
В рационе должны быть антиоксиданты и витамины группы А,С,Е. Стараться не допускать простуды и переохлаждения, это поможет чувствовать себя лучше.

На сегодняшний день медики не могут полностью устранить невральную амиотрофию. Если получилось так, что болезнь затронула пациента, то не стоит переживать. Нужно будет слегка поменять свой образ жизни. Чем раньше обратиться к врачу, тем быстрее можно избавиться от симптомов заболевания.
Лечение должно быть назначено только квалификационным врачом. Заниматься самолечением не стоит, иначе последствий не избежать. При первых симптомах лучше всего посетить сразу же медицинское учреждение. Врач направит на определенные исследования и только после этого назначит комплексную терапию. Если невральная амиотрофия будет запущенно, то серьезных последствий не избежать.
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.



- (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
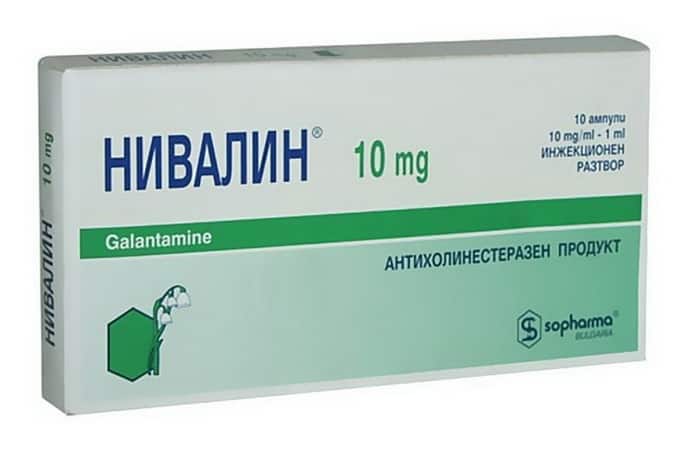
- (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Наследственное заболевание. Основной тип передачи – аутосомно-доминантный (с пенетрантностью патологического гена около 83%), реже – аутосомно-рецессивный.
Морфологическую основу болезни составляют дегенеративные изменения главным образом в периферических нервах и нервных корешках, касающиеся как осевых цилиндров, так и миелиновой оболочки. Иногда наблюдаются гипертрофические явления в интерстициальной ткани. Изменения в мышцах носят преимущественно неврогенный характер, отмечается атрофия отдельных групп мышечных волокон; в неатрофированных мышечных волокнах структурные изменения отсутствуют. По мере прогрессирования заболевания появляются гиперплазия интерстициальной соединительной ткани, изменения в мышечных волокнах – их гиалинизация, центральное смещение ядер сарколеммы, гипертрофия некоторых волокон. В более поздних стадиях болезни отмечаются гиалиновая дегенерация, распад мышечных волокон. Наряду с этим в ряде случаев отмечены изменения в спинном мозге. Они складываются из атрофии клеток передних рогов, главным образом в поясничной и шейной части спинного мозга, и различной степени поражения проводниковых систем, характерного для наследственной атаксии Фридрейха.
Историческая справка
Впервые официальное описание заболевания появилось в 1886 году в журнале La revue de medicine. Одним из авторов был Жан Мартен Шарко (Франция), которому на тот момент уже шел 61 год и он был известным медиком страны. Вторым автором, участвующим в описании заболевания и его симптомов, был его ученик, который практиковал всего лишь 3 года и звали его Пьер Мари.
Вторая статья была написана совсем молодым неврологом Говардом Генри Тутом (Англия), он только год назад получил приставку к своему имени – MD. И с того момента, заболевание получило название, состоящее из фамилий трех врачей.
Понятное дело, что это были не первые публикации о перонеальной мышечной атрофии. Еще ранее в записях Рудольфа Вирхова (1855 год) присутствует описание заболевания, но не столь подробное и без признаков.
Лечение
Основная причина развития невральной амиотрофии является генетическая предрасположенность. Вылечить полностью заболевание не получится, но замедлить прогрессирование патологии можно. Существует ряд методов, которые помогают устранить симптоматику болезни.
Применяются следующие способы терапии:
- Физиотерапевтические процедуры;
- Лечебные физические упражнения;
- Лекарственные средства;
- Массаж;
- Хирургические вмешательство.=
Если соблюдать все рекомендации врача, то это поможет устранить симптомы в короткие сроки. Главное, вовремя обратиться к врачу для того чтобы он назначил лечение. Если игнорировать первые проявления невральной амиотрофии то это может привести к серьезным последствиям.

Назначаются следующие медикаменты для облегчения состояния больного:
- курс специальных витаминных добавок, чтобы улучшить питание мышечных волокон;
- Нивалин и Прозерин помогает ускорить нервную проводимость;
- Никотиновая кислота, Галидор приводят в норму кровоснабжение.
Лечение подбирается под каждого пациента индивидуально. Всё зависит от того на какой стадии развития заболевание. Для того чтобы больной почувствовал себя легче, нужно будет применять определенные методы лечения. Как правило, одних медикаментов бывает недостаточно, поэтому необходимо сочетать с другими способами терапии. Лучше всего обращаться к врачу при первых проявлений патологии.
Массажные процедуры хорошо помогают справиться с признаками невральной амиотрофии. Массаж делается, как руками, так и аппаратом и происходит воздействие на тело. Массажные процедуры захватывают кожу, мышцы, сосуды, нервы. Если использовать несколько видов массажей, то это прежде всего поможет расслабиться. Можно добиться тонизирующего эффекта, болеутоляющего и так далее.
Важно также соблюдать правильное питание, ведь это играет большую роль в терапии. Невральная амиотрофия приводит к утрате мышечной массе, поэтому придется, чтобы рацион был сбалансированным
Необходимо, чтобы была белковая диета и в питании присутствовали аминокислоты. Может быть так, что организм не усваивает белок. Тогда придется искать причину почему, так происходит.
Должны обязательно проводится физические упражнения. Перед тем, как их выполнять врач должен индивидуально подбирать. При заболевании происходит отказ определенных мышц и другие берут на себя работоспособность пораженных участков. Если неправильно будут назначены упражнения, то от лечения не будет никакого эффекта. Только врач должен подбирать под каждого больного определенные физические нагрузки.
Взрослым людям часто нужна психологическая помощь при заболевании. Когда происходит развитие патологии, то у них могут возникать нехорошие мысли. На подобии того, что они приносят только лишние проблемы своим родным из-за болезни.

В детском возрасте нужна помощь только в том, случае, если необходимо развиваться интеллектуально. Ведь, как правило, мышцы также учувствуют в развитии головного мозга. Для того чтобы не допустить отставание ребенка нужно применять специальные интеллектуальные упражнения.
Существует ряд продуктов, которые запрещается употреблять при заболевании. Отказаться необходимо от алкоголя и энергетиков. Сладкие напитки и кофе также нужно исключить из рациона. Употребление соли необходимо сократить, так как это провоцирует развитие заболевания. Продукты быстрого приготовления и фаст-фуд приводят к разрушению мышечных волокон. Патология называется Шарко-Мари-Тута или наследственная моторно-сенсорная нейропатия.
1.2.2. Невральная амиотрофия шарко-мари-тута.
Частота 1:500000 населения. Наследуется по аутосомно-доминантному, аутосомно-рецессивному сцепленному с Х-хромосомой типу. Обнаруживается сегментарная демиелинизация внервах, в мышцах — денервация с явлениями «пучковой» атрофии мышечных волокон. КЛИНИКА. Первые признаки заболевания чаще проявляются в 15 — 30 лет, реже в дошкольном возрасте. Характерными симптомами являются мышечная слабость, патологическая утомляемость в дистальных отделах ног
. Больные быстро устают при длительном стоянии на одном месте и нередко для уменьшения утомления в мышцах прибегают к ходьбе на месте («симптом топтания «). Реже заболевание начинается с чувствительных расстройств — болей, парестезий, ощущения ползания мурашек. Атрофии первоначально развиваются в мышцах голеней и стоп. Мышечные атрофии, как правило, симметричные. Поражается перонеальная группа мышц и передняя большеберцовая мышца. Вследствие атрофий ноги резко сужаются в дистальных отделах и приобретаютформу «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся«выеденными» с высоким сводом . Парез стоп изменяет походку больных. Они ходят, высоко поднимая ноги; ходьба на пятках невозможна. Атрофии в дистальных отделах рук — мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются позже. Ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлекс с трех-двуглавой мышц плеча длительное время остаются сохранными. Чувствительные расстройства объективно определяются нарушениями поверхностной чувствительности по периферическому типу(тип «перчаток» и «носков»). Часто имеются вегетативно-трофические нарушения — гипергидроз стоп и кистей, гиперемия кистей и стоп. Интеллект обычно сохранен.
Течение медленно прогрессирующее. Прогноз благоприятен.
Диагностика. Тип наследования, атрофии дистальных отделов конечностей, расстройства чувствительности по полиневритическому типу, медленное прогрессирующее течение, результаты электромиографии (снижение скорости проведения по периферическим нервам), биопсия нервов.
ЛЕЧЕНИЕ ПМД. Применяют витамины
группы В, С Е, а такжеАТФ, церебролизин, ноотропил, энцефабол, фосфаден, карнитина хлорид, метионин, лецитин, глутаминовая кислота, ретаболил. Положительный эффект даютантихолинэстеразные препараты (прозерин, местинон, галантамин). Показаны средства, улучшающие микроциркуляцию:никотиновая кислота, трентал, пармидин . Наряду с медикаментозной терапией применяютЛФК, массаж , электрофорез лекарственных средств (прозерин, кальция хлорид), диадинамические токи, синусоидальные модулированные токи, электростимуляция, ультразвук, озокерит, грязевые аппликации, радоновые, хвойные, сульфидные и сероводородные ванны, оксигенобаротерапия. Показаноортопедическое лечение при контрактурах конечностей, умеренной деформации позвоночника и асимметричном укорочении конечностей.
13.2. Семейная атаксия фридрейха.
Наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковыхстолбов спинного мозга . Тип наследования аутосомно-рецессивный с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
КЛИНИКА. Начало заболевания относится к 6 — 15-летнему возрасту. Первым симптомом болезни является неустойчивая походка
. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на руки и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Нарушается почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередко патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен. Заболевание медленно прогрессирует. Средняя продолжительность жизни 10 — 15 лет с момента его развития.
Диагностика. Заболевание распознается на основании характерных симптомов — деформаций стоп по типу стопы Фридрейха
(высокий свод, экстензия основных фаланг пальцев стопы, флексия концевых фаланг), позвоночника, поражения миокарда, эндокринных расстройств.
ЛЕЧЕНИЕ. Применяются симптоматические средства: общеукрепляющие препараты, ЛФК, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Лечение БАС
 Увы, болезнь Шарко является неизлечимой. То есть сегодня еще нет ни одного варианта замедлить (либо приостановить) прогрессирование заболевания на длительное время.
Увы, болезнь Шарко является неизлечимой. То есть сегодня еще нет ни одного варианта замедлить (либо приостановить) прогрессирование заболевания на длительное время.
На сегодняшний день разработан только единственный препарат, достоверно продлевающий жизнь пациентам с боковым амиотрофическим склерозом. Это препарат, который предотвращает выброс глутамата — Рилузол. Его нужно постоянно принимать по 100 мг ежедневно. Но Рилузол увеличивает приблизительно продолжительность жизни лишь на три месяца. Как правило, его назначают пациентам, у которых болезнь проходит меньше 5 лет, с самостоятельным дыханием. Во время его приема необходимо учитывать побочное явление в форме лекарственного гепатита. Потому люди, которые используют Рилузол, обязаны раз в три месяца выполнять обследование печени.
Всем пациентам, страдающим болезнью Шарко, назначают симптоматическое лечение. Его основная задача – минимизировать необходимость в постороннем уходе, улучшить качество жизни, облегчить страдания.
Симптоматическая терапия необходима при таких нарушениях:
- для повышения мышечного метаболизма – Левокарнитин, Карнитин (Элькар), Берлитион (Липоевая кислота, Эспа-Липон);
- при слюнотечении — Амитриптилин в таблетках, Атропин закапывать в рот, применение портативных отсосов, механическая чистка полости рта, облучение слюнных желез, уколы ботулотоксина в слюнные железы;
- при крампи и фасцикуляциях – Сирдалуд (Тизанидин), Баклофен (Лиорезал), Карбамазепин (Финлепсин);
- для повышения обменных процессов в нейронах — витамины В (Комбилипен, Мильгамма и т.д.);
- при депрессиях – Амитриптилин, Сертралин (Золофт), Флуоксетин (Прозак).
Большинство симптомов при БАС нуждаются в немедикаментозных способах воздействия.
Если у пациента появляются сложности с проглатыванием пищи, то нужно переходить на питание перемолотыми и протертыми продуктами, использовать полужидкие каши, пюре, суфле. После любого приема пищи необходимо выполнять санацию ротовой полости.
Когда прием пищи настолько осложняется, что пациент вынужден пережевывать порцию пищи больше получаса, если в день не может выпить больше литра жидкости или при прогрессирующем снижении веса больше чем на 3% в месяц, то нужно задуматься о выполнении эндоскопической чрезкожной гастростомии, операции после которой еда проходит в организм с помощью трубки, которую вводят в область живота.
Когда пациент не согласен на выполнение этой операции, а прием пищи полностью невозможен, то нужно переходить на зондовое питание (в желудок через рот устанавливают зонд, куда вливают еду). Можно использовать ректальный (через прямую кишку) или парентеральный (внутривенный) прием пищи. Эти способы позволяют пациентам не умереть от голода.
Чтобы не допустить тромбоза вен конечностей пациент обязан использовать эластичные бинты. При развитии инфекционных осложнений назначаются антибиотики.
Частично двигательные симптомы можно откорректировать с помощью специальных ортопедических устройств. Для поддержания ходьбы используют ходунки, трости, ортопедическую обувь, а через время и коляски. При отвисании головы используют жесткие или полужесткие головодержатели. На поздних этапах заболевания пациенту требуется функциональная кровать.
Одним из самых тяжелых симптомов при боковом амиотрофическом склерозе считается нарушение дыхания. Если показатели количества кислорода в крови снижаются до критических и появляется ярко выраженная дыхательная недостаточность, то в этом случае показано применение устройств неинвазивной периодической вентиляции. Их можно использовать пациентам и дома, но из-за дороговизны, они малодоступны.
 Когда потребность во вмешательстве в дыхательный процесс составляет более 18 часов в сутки, то пациенту назначается искусственная вентиляция легких и трахеостомия. Время, когда пациент начинает нуждаться в трахеостомии критическое, поскольку говорит о приближающейся смерти. С точки зрения медицинской этики вопрос перевода пациента на трахеостомию довольно сложный. Так как данная процедура сохраняет жизнь на определенное время, но также и продлевает мучения, так как пациенты с болезнью Шарко довольно продолжительное время находятся в рассудке.
Когда потребность во вмешательстве в дыхательный процесс составляет более 18 часов в сутки, то пациенту назначается искусственная вентиляция легких и трахеостомия. Время, когда пациент начинает нуждаться в трахеостомии критическое, поскольку говорит о приближающейся смерти. С точки зрения медицинской этики вопрос перевода пациента на трахеостомию довольно сложный. Так как данная процедура сохраняет жизнь на определенное время, но также и продлевает мучения, так как пациенты с болезнью Шарко довольно продолжительное время находятся в рассудке.
Болезнь Шарко – это тяжелое неврологическое состояние, которое сегодня не оставляет почти никаких шансов для пациентов
Довольно важно правильно установить диагноз. Эффективного лечения этой болезни пока нет
Весь комплекс мероприятий, как социальных, так и медицинских, выполняемых в случае БАС, обязан направляться на организацию максимально полноценной жизни пациента.
В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
При описываемом заболевании у человека первым делом берет начало формирование схожих мышечных некрозов в нижних конечностей. Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
Стоит знать, что пагубные поражения никогда не поражают мышцы шейного периметра, туловища и плечевого района. Помимо вышеперечисленных симптомов, у больного могут появиться следующие проблемы:
- Слабое подергивание.
- Гипертрофия компенсаторной природы мышечных областей.
- Сенсорные нарушения.
- Возможность появления цианоза и отечности на кожном покрове.
Для ШМТ типично медленное развитие симптоматики. Период развития процесса уменьшения в размерах ног и рук может занять до десяти лет. Даже при таких серьёзных деформациях тела больной долгое время способен сохранять работоспособность и нормально выполнять различные бытовые обязанности. Ускорителями симптомов могут стать следующие факторы:
- Попадание в тело инфекционных агентов.
- Долгое пребывание на холоде.
- Травмирования головы.
- Повреждения спины и спинного мозга.
- Нехватка витаминов в организме.